









1
Mitonuclear co-introgression opposes genetic differentiation between
1
phenotypically divergent songbirds
2
3
Ellen Nikelski
1, *
, Alexander S. Rubtsov
2
, and Darren Irwin
1
4
5
1
Department of Zoology, and Biodiversity Research Centre, 6270 University Blvd., University of British Columbia,
6
Vancouver, BC, Canada
7
2
State Darwin Museum, Moscow, Russia
8
*
Present address: Department of Ecology and Evolutionary Biology, University of Toronto, Toronto, ON, Canada
9
10
Running Title:
11
Co-introgression opposes differentiation
12
13
Corresponding Author:
14
Ellen Nikelski, Department of Ecology and Evolutionary Biology, University of Toronto,
15
Toronto, ON, Canada. Email: ellen.nikelski@mail.utoronto.ca
16
17
Keywords:
18
mitonuclear co-introgression, mtDNA, mitonuclear gene, genetic differentiation, chromosome Z,
19
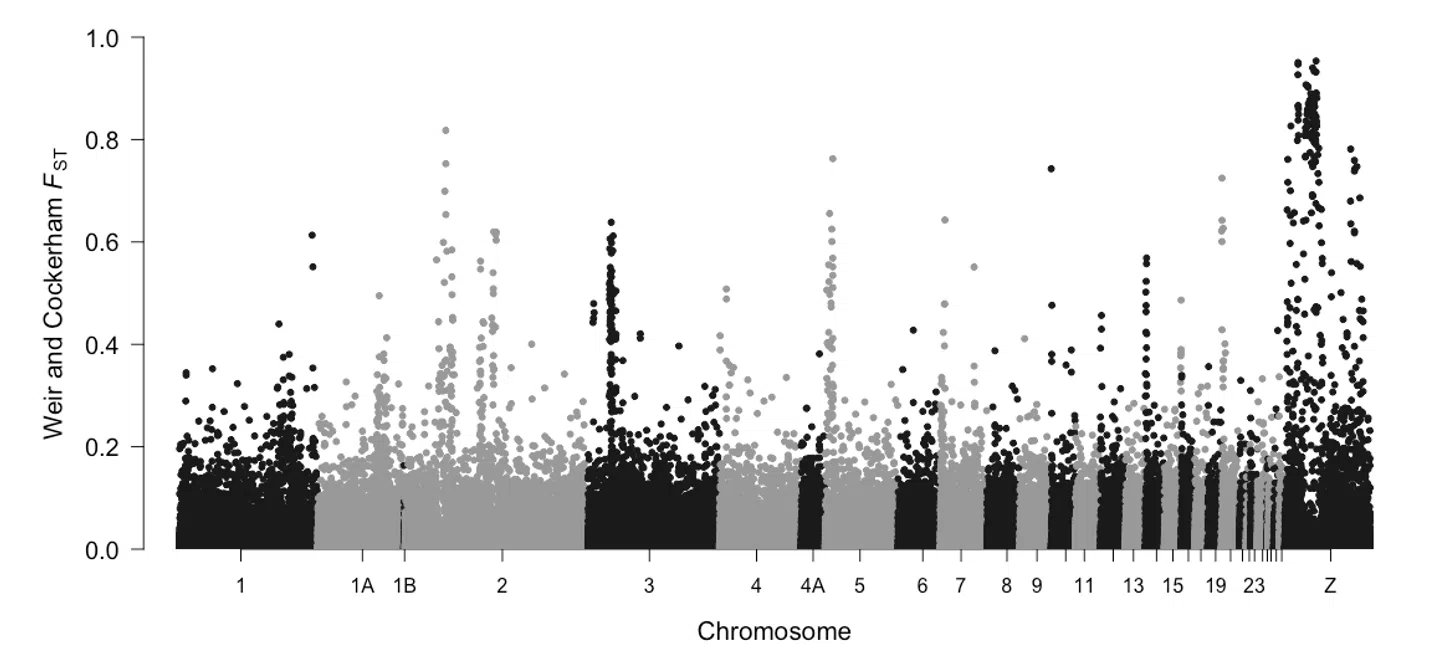
Aves
20
21
22
23
24
25
26
27
28
29
30
31
.CC-BY-NC-ND 4.0 International licenseavailable under a
(which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprintthis version posted August 8, 2021. ; https://doi.org/10.1101/2021.08.08.455564doi: bioRxiv preprint

2
Abstract
32
Comparisons of genomic variation among closely related species often show more differentiation
33
in mitochondrial DNA (mtDNA) and sex chromosomes than in autosomes, a pattern expected
34
due to the relative effective population sizes of these genomic components. Differential
35
introgression can cause some species pairs to deviate dramatically from this pattern. The
36
yellowhammer (Emberiza citrinella) and the pine bunting (E. leucocephalos) are hybridizing
37
avian sister species that differ greatly in appearance but show no mtDNA differentiation. This
38
discordance might be explained by mtDNA introgression—a process that can select for co-
39
introgression at nuclear genes with mitochondrial functions (mitonuclear genes). We investigated
40
genome-wide nuclear differentiation between yellowhammers and pine buntings and compared it
41
to what was seen previously in the mitochondrial genome. We found clear nuclear differentiation
42
that was highly heterogeneous across the genome, with a particularly wide differentiation peak
43
on the sex chromosome Z. We further tested for preferential introgression of mitonuclear genes
44
and detected evidence for such biased introgression in yellowhammers. Mitonuclear co-
45
introgression can remove post-zygotic incompatibilities between species and may contribute to
46
the continued hybridization between yellowhammers and pine buntings despite their clear
47
morphological and genetic differences. As such, our results highlight the potential ramifications
48
of co-introgression in species evolution.
49
50
Introduction
51
Evolution in eukaryotes is shaped by changes in multiple genomic components that differ
52
in their modes of inheritance: mitochondrial DNA (mtDNA) is usually inherited through the
53
.CC-BY-NC-ND 4.0 International licenseavailable under a
(which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprintthis version posted August 8, 2021. ; https://doi.org/10.1101/2021.08.08.455564doi: bioRxiv preprint

3
matrilineal line, autosomes are inherited through both parental lines and sex chromosomes are
54
inherited differentially depending on the sex of both parent and offspring (Avise, 2000). There is
55
often much variation among these genomic components in the degree of genetic differentiation
56
between related populations or species (reviewed in Coyne & Orr, 2004; reviewed in Price,
57
2008), suggesting that their dynamics differ during the process of speciation of a single species
58
into two or more. This variation can arise through differences in both the rate at which specific
59
DNA sequences evolve and the degree to which different components contribute towards genetic
60
incompatibilities that reduce gene flow between populations. A common pattern observed
61
between speciating taxa is clear differentiation in mtDNA (eg. Hebert et al. 2004; Kerr et al.
62
2007), moderate differentiation in sex chromosomes (eg. Thornton & Long, 2002; Borge et al.
63
2005; Lu & Wu, 2005; Harr, 2006; Ruegg et al. 2014; Sackton et al. 2014), and comparatively
64
modest differentiation across autosomes (Harr, 2006; Nadeau et al. 2012; Irwin et al. 2018).
65
Measures of mtDNA differentiation are often used to identify and classify genetically
66
distinct populations (eg. Hebert et al. 2004; Kerr et al. 2007) and to infer their histories (Moore,
67
1995; Zink & Barrowclough, 2008). Due to its uniparental inheritance, mtDNA has one quarter
68
the effective population size and coalescence time of autosomal nuclear DNA (Moore, 1995).
69
This characteristic combined with mtDNA’s relatively high mutation rate (Lynch et al. 2006)
70
mean that genetic differences arise and fix relatively quickly, creating patterns of clear mtDNA
71
differentiation between recently diverged populations.
72
Sex chromosomes are another genomic region that often shows higher between-
73
population genetic differentiation compared to autosomes between speciating taxa, in both Z/W
74
(Borge et al. 2005; Ruegg et al. 2014; Sackton et al. 2014) and X/Y systems (Thorton & Long,
75
2002; Lu & Wu, 2005; Harr, 2006). To explain this “faster Z/X effect,” researchers have noted
76
.CC-BY-NC-ND 4.0 International licenseavailable under a
(which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprintthis version posted August 8, 2021. ; https://doi.org/10.1101/2021.08.08.455564doi: bioRxiv preprint

4
that, because beneficial recessive mutations on the Z or X chromosome are immediately exposed
77
to selective forces in the heterogametic sex, fixation of these mutations should proceed faster
78
than if the mutations appeared on autosomes (reviewed in Meisel & Connallon, 2013; Irwin,
79
2018). Also contributing to genetic differentiation on the Z and X chromosomes are the lower
80
effective population sizes of these chromosomes compared to autosomes (Mank et al. 2010;
81
reviewed in Irwin, 2018). A lower effective population size allows for the fixation of a greater
82
number of slightly deleterious mutations due to less effective purifying selection and a larger role
83
of genetic drift. It is likely that both forces—the faster Z/X effect and less effective purifying
84
selection—contribute to the moderate amount of genetic differentiation seen between the sex
85
chromosomes of diverging taxa (Thorton & Long, 2002; Borge et al. 2005; Lu & Wu, 2005;
86
Harr, 2006; Ruegg et al. 2014; Sackton et al. 2014).
87
Differentiation across autosomes, which tends to be lower than on mtDNA and sex
88
chromosomes, can be highly heterogeneous. In fact, many researchers report “islands of
89
differentiation” on autosomes where peaks of high relative differentiation are found against a
90
background of low relative differentiation (e.g., Harr, 2006; Nadeau et al. 2012; Hejase et al.
91
2020). Explanations for these “islands” usually invoke reduced gene flow (reviewed in Wu,
92
2001) and/or repeated bouts of selection (Cruickshank and Hahn, 2014; Irwin et al. 2018). In the
93
former scenario, differentiation peaks are hypothesized to house the loci responsible for
94
reproductive barriers between interacting taxa and, as a result, they are resistant to the gene flow
95
that homogenizes the rest of the nuclear genome. In contrast, explanations invoking repeated
96
selection hypothesize that differentiation islands are areas of the genome that experienced
97
repeated reductions in genetic diversity as a result of selection or selective sweeps in both
98
ancestral and daughter populations.
99
.CC-BY-NC-ND 4.0 International licenseavailable under a
(which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprintthis version posted August 8, 2021. ; https://doi.org/10.1101/2021.08.08.455564doi: bioRxiv preprint

5
Despite the general patterns of differentiation discussed above, an increasing number of
100
studies report remarkably low differentiation between populations at what are normally highly
101
divergent genetic components when compared to other genetic regions or observable phenotypes
102
(e.g., Irwin et al. 2009; Yannic et al. 2010; Bryson et al. 2012). In a number of cases, mtDNA
103
shows dramatically low differentiation when compared to differentiation of the nuclear genome,
104
a pattern referred to as “mitonuclear discordance” (reviewed in Toews & Brelsford, 2012).
105
Discordance between marker types may be explained by hybridization and introgression between
106
populations, perhaps due to a selective advantage of the introgressing genetic region. For
107
example, Hulsey et al. (2016) documented low mtDNA differentiation—likely due to
108
introgression—and clear differentiation in nuclear DNA (nucDNA) between two hybridizing
109
cichlid species (Hulsey & García de León, 2013). The researchers further reported high mtDNA
110
differentiation between isolated populations of cichlids at genetic sites associated with thermal
111
tolerance and a significant correlation between mtDNA divergence and water temperature
112
(Hulsey et al. 2016). Altogether, these results suggest that mtDNA introgression produced the
113
discordance seen between marker types and that this outcome was potentially driven by adaptive
114
selection for tolerance of extreme water temperatures.
115
The hypothesis of adaptive introgression increases in complexity if we consider the
116
potential for coevolution between genomic components. Research investigating coevolution
117
between mitochondrial and nuclear genomes is relatively novel as mtDNA was often treated as a
118
neutral marker in past evolutionary research (Avise, 2000). Nevertheless, recent empirical and
119
theoretical work has provided greater context regarding how mitonuclear coevolution may
120
influence the progression of differentiation and speciation between taxa (Hill, 2019).
121
.CC-BY-NC-ND 4.0 International licenseavailable under a
(which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprintthis version posted August 8, 2021. ; https://doi.org/10.1101/2021.08.08.455564doi: bioRxiv preprint